
Text
INDEX
INTRODUCTION
The administration of a single high dose or of multiple doses of MDMA ("ecstasy") to rats produces long-term alterations in different serotonergic parameters. A marked reduction in the content of 5-HT and its metabolite 5-HIAA along with a decrease in tryptophan hydroxylase activity in various brain regions has been described (Stone et al., 1986![]() ; Schmidt, 1987
; Schmidt, 1987![]() ; Schmidt and Taylor, 1987
; Schmidt and Taylor, 1987![]() ). MDMA has also been shown to reduce the number of [3H]paroxetine-labeled 5-HT transporters (Aguirre et al., 1995
). MDMA has also been shown to reduce the number of [3H]paroxetine-labeled 5-HT transporters (Aguirre et al., 1995![]() ; Battaglia et al., 1987
; Battaglia et al., 1987![]() ) as well as the synaptosomal uptake of [3H]5-HT (Schmidt, 1987
) as well as the synaptosomal uptake of [3H]5-HT (Schmidt, 1987![]() ). Furthermore, immunocytochemical evidence indicates that MDMA selectively damages fine axon terminals that arise from cell bodies located in the dorsal raphe nucleus of the midbrain which remain spared (O'Hearn et al., 1988
). Furthermore, immunocytochemical evidence indicates that MDMA selectively damages fine axon terminals that arise from cell bodies located in the dorsal raphe nucleus of the midbrain which remain spared (O'Hearn et al., 1988![]() ; Slikker et al., 1988
; Slikker et al., 1988![]() ).
).
Although the precise mechanism by which MDMA selectively damages 5-HT axon terminals remains largely unknown, studies from different laboratories have provided evidence for an important role of dopamine in MDMA-induced neurotoxicity. Stone et al. (1988)![]() showed that previous dopamine depletion with the synthesis inhibitor
showed that previous dopamine depletion with the synthesis inhibitor  -methyl-p-tyrosine, or treatment with the dopamine uptake blocker GBR-12909 prevent the long-term reductions of 5-HT induced by MDMA. In an analogous fashion, if dopamine terminals are destroyed with 6-hydroxydopamine, 5-HT terminals remain spared after MDMA (Schmidt et al., 1990b
-methyl-p-tyrosine, or treatment with the dopamine uptake blocker GBR-12909 prevent the long-term reductions of 5-HT induced by MDMA. In an analogous fashion, if dopamine terminals are destroyed with 6-hydroxydopamine, 5-HT terminals remain spared after MDMA (Schmidt et al., 1990b![]() ). It has also been shown that MDMA increases both in vivo dopamine release (Koch and Galloway, 1997
). It has also been shown that MDMA increases both in vivo dopamine release (Koch and Galloway, 1997![]() ; Nash, 1990
; Nash, 1990![]() ) via a carrier-mediated mechanism (Gudelsky and Nash, 1996
) via a carrier-mediated mechanism (Gudelsky and Nash, 1996![]() ; Nash and Brodkin, 1991
; Nash and Brodkin, 1991![]() ) and dopamine synthesis (Nash et al., 1990
) and dopamine synthesis (Nash et al., 1990![]() ) through 5-HT2 receptor stimulation (Huang and Nichols, 1993
) through 5-HT2 receptor stimulation (Huang and Nichols, 1993![]() ), although the latter effect only becomes significant during states of high serotonergic and dopaminergic transmission (Schmidt et al., 1992b
), although the latter effect only becomes significant during states of high serotonergic and dopaminergic transmission (Schmidt et al., 1992b![]() ). Microdialysis studies have shown that blockade of striatal 5-HT2 receptors reduces significantly the efflux of endogenous dopamine (Schmidt et al., 1994
). Microdialysis studies have shown that blockade of striatal 5-HT2 receptors reduces significantly the efflux of endogenous dopamine (Schmidt et al., 1994![]() ) whereas the activation of these receptors with the agonist R-DOI potentiates MDMA-induced dopamine release and serotonergic toxicity (Gudelsky et al., 1994
) whereas the activation of these receptors with the agonist R-DOI potentiates MDMA-induced dopamine release and serotonergic toxicity (Gudelsky et al., 1994![]() ). Other studies have demonstrated that selective 5-HT2 receptor antagonists protect against the neurotoxicity induced by MDMA (Schmidt et al., 1990a
). Other studies have demonstrated that selective 5-HT2 receptor antagonists protect against the neurotoxicity induced by MDMA (Schmidt et al., 1990a![]() ,b
,b![]() ); however, L-DOPA coadministration abolishes the protective effect of 5-HT2 receptor antagonists (Schmidt et al., 1991b
); however, L-DOPA coadministration abolishes the protective effect of 5-HT2 receptor antagonists (Schmidt et al., 1991b![]() , 1992a
, 1992a![]() ). Furthermore, pretreatment with L-DOPA potentiates the long-term serotonergic deficits induced by MDMA (Schmidt et al., 1991a
). Furthermore, pretreatment with L-DOPA potentiates the long-term serotonergic deficits induced by MDMA (Schmidt et al., 1991a![]() ) and there appears to exist a linear correlation between the acute increase of extracellular dopamine and the extent of serotonergic toxicity (Nash and Nichols, 1991
) and there appears to exist a linear correlation between the acute increase of extracellular dopamine and the extent of serotonergic toxicity (Nash and Nichols, 1991![]() ). It has been recently suggested that dopamine would then be transported into the 5-HT terminal where it is deaminated by MAO-B resulting in an elevated intracellular level of hydrogen peroxide that would lead to an extensive lipid peroxidation in the 5-HT terminal membrane and a subsequent neuronal degeneration (Sprague and Nichols, 1995a
). It has been recently suggested that dopamine would then be transported into the 5-HT terminal where it is deaminated by MAO-B resulting in an elevated intracellular level of hydrogen peroxide that would lead to an extensive lipid peroxidation in the 5-HT terminal membrane and a subsequent neuronal degeneration (Sprague and Nichols, 1995a![]() ,b
,b![]() ). Results from other laboratories support the hypothesis that MDMA produces acute inactivation of tryptophan hydroxylase and neurotoxicity of 5-HT neurons because of oxidative events probably associated with
dopamine metabolism (Cadet et al., 1995
). Results from other laboratories support the hypothesis that MDMA produces acute inactivation of tryptophan hydroxylase and neurotoxicity of 5-HT neurons because of oxidative events probably associated with
dopamine metabolism (Cadet et al., 1995![]() ; Colado and Green, 1995
; Colado and Green, 1995![]() ; Gudelsky, 1996
; Gudelsky, 1996![]() ; Gudelsky and Yamamoto, 1994
; Gudelsky and Yamamoto, 1994![]() ).
).
Although there are numerous studies related to the neurochemical consequences of MDMA on the 5-HT system of adult rats, studies on the effects of MDMA administration to rats at early postnatal ages are scarce. St. Omer et al. (1991)![]() found that MDMA administered during gestation had no effect on the postnatal neurochemical development of the serotonergic system of the rat. Similarly, Broening et al. (1994)
found that MDMA administered during gestation had no effect on the postnatal neurochemical development of the serotonergic system of the rat. Similarly, Broening et al. (1994)![]() showed that rats do not develop their sensitivity to the long-term neurochemical deficits induced by MDMA until postnatal day 40.
showed that rats do not develop their sensitivity to the long-term neurochemical deficits induced by MDMA until postnatal day 40.
The first aim of our study was to assess the effect of perinatal administration of MDMA and to determine the period for the onset of susceptibility to the neurotoxic effects of MDMA in the rat pups exposed to this drug. We also tried to ascertain why at early postnatal ages MDMA does not produce, as in adult rats, a long-term reduction in brain 5-HT content and 5-HT transporter density. The study was focused on brain 5-HT2 receptors and dopamine content, which appear to play a major role in the neurotoxicity induced by MDMA. Adult rats with a previous dopamine depletion were comparatively studied. As the hyperthermia induced by MDMA appears to contribute to its neurotoxic effects (Malberg et al., 1996![]() ), the correlation of the thermal response to MDMA with the neurotoxic effect was also analyzed.
), the correlation of the thermal response to MDMA with the neurotoxic effect was also analyzed.
METHODS
Animals and treatments. Pregnant female Wistar rats (270-290 g) were individually housed in plastic cages in a temperature controlled room (22 ± 1°C) and maintained on a 12-hr light-dark cycle with free access to food and water. MDMA (20 mg/kg s.c.) was given every other day to rat dams from embryonic day 6 (E6) to E20. The rat pups were killed at PND15. For the rest of the experiments with the rat pups, upon delivery (PND0), offspring in each litter was randomly culled to eight pups. At PND20, litters were weaned and male pups were housed five to a cage. Animals received saline (control group) or a single dose of MDMA (20 mg/kg s.c.) at different postnatal ages: PND14, 21, 28 and 35. In a different set of experiments, 21-day-old rats were distributed into six groups that received: 1) saline followed by saline, 2) saline followed by a single dose of MDMA (20 mg/kg s.c.), 3) L-DOPA (80 mg/kg s.c.) 15 min before saline, 4) L-DOPA 15 min before MDMA, 5) R-DOI (0.5 mg/kg s.c.) followed by saline and 6) R-DOI followed by MDMA. L-DOPA was always administered in combination with the peripheral decarboxylase inhibitor benserazide (20 mg/kg s.c.) and the doses of MDMA used refer to the hydrochloride.
Adult rats (ca. 3-mo-old) received desipramine-HCl (25 mg/kg i.p.) 1 hr before the injection of 6-hydroxydopamine-HBr (100 µg/10 µl expressed as free base) or saline, containing ascorbic acid 0.1%, in each lateral ventricle. Lesions were performed under pentobarbital anaesthesia (50 mg/kg i.p.). Eight days later, sham- and 6-hydroxydopamine-lesioned rats, were treated with saline, MDMA, L-DOPA or L-DOPA + MDMA as above indicated. In all cases animals were killed 7 days after the different drug treatments, their brains were rapidly removed and placed on ice. The appropriate brain regions were dissected free, frozen on dry ice and stored at -80°C until chromatographic and binding studies were performed.Biochemical measurements.
The concentrations of 5-HT, 5-HIAA, dopamine, DOPAC and HVA in the brain regions examined, were determined by high-performance liquid chromatography with electrochemical detection as previously described (Pérez-Otaño et al., 1991![]() ).
).
5-HT transporter density.
[3H]paroxetine binding studies to the 5-HT transporter were performed according to the procedure described by Marcusson et al. (1988)![]() , with minor modifications. The brain regions studied were homogenized in 15 ml of ice-cold buffer (Tris-HCl 50 mM, 120 mM NaCl, 5 mM KCl, pH 7.4) and centrifuged at 48,000 × g for 10 min at 4°C. The pellet was resuspended in buffer and incubated at 37°C for 10 min. After a second centrifugation in the same conditions the resultant
pellet was resuspended in buffer (1.5 mg tissue/400 µl buffer). The incubation mixture contained 400 µl of tissue suspension, 200 µl of increasing concentrations of [3H] paroxetine (0.02-0.4 nM) and 1.4 ml of incubation buffer in the absence and presence of fluoxetine 10 µM. Tubes were incubated for 60 min at 22°C. After rapid filtering through GF/C Whatman filters using a 24-well cell harvester, the filters were rinsed with 4 × 5 ml of ice cold buffer and placed in vials containing 4 ml of liquid scintillation cocktail (Biogreen3, Scharlau). All the determinations were carried out in duplicate. Data were subjected to Scatchard analysis to determine the number of binding sites (Bmax: fmol/mg of protein) and the dissociation constant
(Kd: nM).
, with minor modifications. The brain regions studied were homogenized in 15 ml of ice-cold buffer (Tris-HCl 50 mM, 120 mM NaCl, 5 mM KCl, pH 7.4) and centrifuged at 48,000 × g for 10 min at 4°C. The pellet was resuspended in buffer and incubated at 37°C for 10 min. After a second centrifugation in the same conditions the resultant
pellet was resuspended in buffer (1.5 mg tissue/400 µl buffer). The incubation mixture contained 400 µl of tissue suspension, 200 µl of increasing concentrations of [3H] paroxetine (0.02-0.4 nM) and 1.4 ml of incubation buffer in the absence and presence of fluoxetine 10 µM. Tubes were incubated for 60 min at 22°C. After rapid filtering through GF/C Whatman filters using a 24-well cell harvester, the filters were rinsed with 4 × 5 ml of ice cold buffer and placed in vials containing 4 ml of liquid scintillation cocktail (Biogreen3, Scharlau). All the determinations were carried out in duplicate. Data were subjected to Scatchard analysis to determine the number of binding sites (Bmax: fmol/mg of protein) and the dissociation constant
(Kd: nM).
Temperature measurements. The rectal temperature of the rats was measured at an ambient temperature of 22 ± 1°C with a lubricated digital thermometer probe (pb 0331, Panlab, Barcelona, Spain) inserted 3 cm into the rectum, the rat being lightly restrained by holding in the hand. Temperature was recorded at PND21, before any drug treatment (MDMA alone or in combination with L-DOPA or R-DOI) and thereafter every 30 min up to 240 min. Probes were re-inserted from time to time and maintained until the temperature stabilized. The number of animals per group are detailed in the figure legend.
Drugs. The sources of the drugs used were as follows: MDMA-HCl was either from Sigma (UK) or was a gift from the "Servicio de Restricción de Estupefacientes" (Dr. L. Domínguez, Madrid, Spain); [3H]paroxetine (22.5 Ci/mmol) was obtained from New England Nuclear (Boston, MA); 5-HT creatinine sulfate and 5-HIAA were from Sigma (St. Louis, MO); fluoxetine-HCl was generously donated by Eli-Lilly and Co., (Indianapolis, IN); R-DOI and desipramine-HCl were from Research Biochemicals International (Natick, MA); 6-hydroxydopamine hydrobromide was from ICN Biomedicals Inc. (Aurora, OH); L-DOPA and benserazide were from Syntex Latino (Madrid, Spain), all other chemicals were from Merck (Darmstadt, Germany).
Statistical analysis. The effect of perinatal administration of MDMA on 5-HT and 5-HIAA concentrations was compared with the saline (control) treated group using an unpaired Student's t test. Analyses of the differences between multiple treatment groups consisted of analysis of variance followed by Tukey post hoc test. For the rectal temperature analysis, two-way analysis of variance for repeated measures was used to compare treatment groups. Single time point comparisons between groups were made using Tukey's test. Significant differences were defined at P < .05.
RESULTS
Effect of perinatal administration of MDMA on 5-HT and dopamine brain levels and 5-HT tranporter density. The effect on brain 5-HT content of MDMA (20 mg/kg s.c.) at different developmental ages is depicted in table 1. To assess a possible neurotoxic action of MDMA on rat progeny, the drug was first administered to rat dams every other day from E6 to E20 and the rat pups were killed at PND15. This treatment had no effect on the postnatal levels of 5-HT. Similarly, a single injection of MDMA (20 mg/kg s.c.) at PND14 and 21 did not cause any significant reduction of 5-HT. When MDMA was administered at PND28, 5-HT concentration was significantly decreased 7 days later in the hippocampus but not in any other terminal field of the serotonergic system examined (frontal cortex, striatum and hypothalamus). A significant long-term reduction of 5-HT levels in all the brain regions examined was already achieved when MDMA was given at PND35 (table 1). 5-HIAA concentration was always modified in a parallel fashion to that of 5-HT (not shown).
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
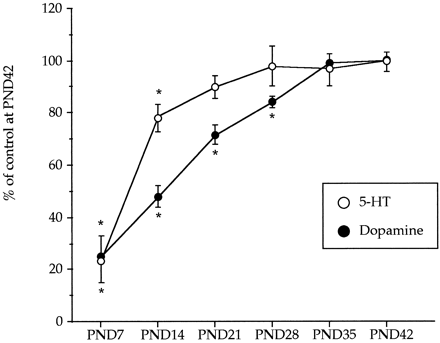
 ) and dopamine (
) and dopamine ( ) during postnatal development. Animals were killed at different postnatal days (PND). All the animals received a saline injection 7 days before sacrifice. Values (n = 10-12) expressed as percentage of striatal monoamine concentrations at PND42. Absolute values from PND21 to PND42 are shown in table
) during postnatal development. Animals were killed at different postnatal days (PND). All the animals received a saline injection 7 days before sacrifice. Values (n = 10-12) expressed as percentage of striatal monoamine concentrations at PND42. Absolute values from PND21 to PND42 are shown in table Effect of combined treatments of MDMA and R-DOI or L-DOPA on brain 5-HT levels and 5-HT transporter density in rat pups. The combined administration of R-DOI and MDMA at PND21 did not result in altered levels of 5-HT 7 days later. However, the administration of L-DOPA (80 mg/kg s.c.) 15 min before a single injection of MDMA (20 mg/kg s.c.) already caused at PND21 a significant long-term reduction of 5-HT levels in the frontal cortex and in the hippocampus. Although the concentration of 5-HT tended to be lower in the striatum and in the hypothalamus, the reduction did not reach statistical significance (fig. 2).
|

 P < .05 vs. MDMA.
P < .05 vs. MDMA.
|
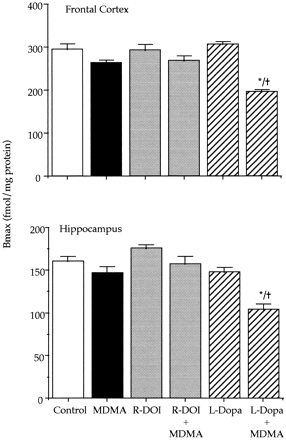
Effect of combined treatment of MDMA and L-DOPA on brain 5-HT levels and 5-HT transporter density in adult rats lesioned with 6-hydroxydopamine. After treatment with 6-hydroxydopamine, the depletion of dopamine was approximately 85% in the striatum and 50% in the hypothalamus. Administration of MDMA (20 mg/kg s.c.) or L-DOPA (80 mg/kg s.c.) to 6-hydroxydopamine-lesioned rats did not produce any change in the concentration of 5-HT (not shown). However, the combination of L-DOPA with MDMA caused a 40 to 60% reduction of 5-HT content in the frontal cortex, hippocampus, striatum and hypothalamus. For example, the 5-HT concentration (pg/mg wet tissue) in the frontal cortex and hippocampus of lesioned rats receiving only MDMA was 418.8 ± 32.2 and 364.8 ± 26.0, respectively (means ± S.E., n = 6). In rats receiving the combined treatment of MDMA and L-DOPA the corresponding values were 159.0 ± 19.8 and 130.0 ± 9.0 (P < .05).
5-HT transporter density (fmol/mg of protein) of 6-hydroxydopamine-lesioned rats was 452.1 ± 23.3 and 268.2 ± 19.4 (means ± S.E., n = 6) in the frontal cortex and in the hippocampus respectively. These values were almost identical to those of the corresponding sham controls. MDMA treatment did not produce any change when administered to 6-hydroxydopamine lesioned rats. By contrast, the combination of L-DOPA and MDMA significantly decreased (P < .05) in the lesioned rats 5-HT transporter density by 40 and 50% in the frontal cortex and in the hippocampus respectively.MDMA induced hyperthermia. MDMA administered at PND21 caused a significant raise of core temperature that lasted approximately 4 hr. When temperature in the MDMA-treated group returned to control values (at 240-min time point), no other temperature measurement was taken in the rest of the groups. The highest raise in temperature for the MDMA treated group (ca. 2°C), was obtained during the first 90 min after drug administration.
L-DOPA administration produced a mild hypothermia (ca. -1.6°C at 120-min time point), although R-DOI had no effect on core temperature of the rat pups when compared to controls. The combination of R-DOI or L-DOPA with MDMA, induced in both cases a significant hyperthermia that lasted beyond the period of measurement (240 min). The highest raise in core temperature was obtained for both groups during the first 90 min (ca. 3° and 2.5°C, respectively) as happened with the MDMA-treated group. The combination of R-DOI and MDMA resulted in temperatures significantly higher than MDMA alone at all time points (P < .05). Significant differences between L-DOPA + MDMA and MDMA-treated groups were obtained only beyond the 120-min time point after injection (P < .05). Results are depicted in figure 4.
|
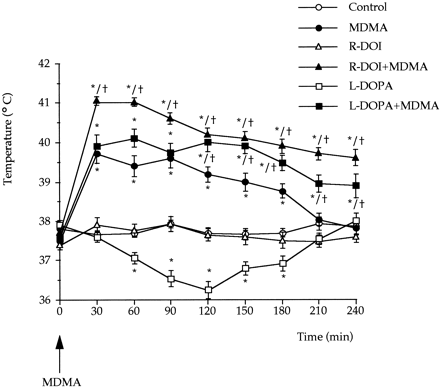
 ); d, L-DOPA 15 min before MDMA (
); d, L-DOPA 15 min before MDMA ( ); e,
(R)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (R-DOI, 0.5 mg/kg s.c.) followed by saline (
); e,
(R)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (R-DOI, 0.5 mg/kg s.c.) followed by saline ( ) and f, R-DOI followed by MDMA (
) and f, R-DOI followed by MDMA ( ). Values are means ± S.E. (n = 7-10). * P < .05 or better vs. control group (saline + saline).
). Values are means ± S.E. (n = 7-10). * P < .05 or better vs. control group (saline + saline). DISCUSSION
Our results indicate that MDMA, administered repeatedly to rat dams during gestation, did not apparently produce any change in the development of the serotonergic system of the offspring. In agreement with previous studies, rat pups were also resistant to the neurotoxic effects of a single dose of MDMA over the first 4 wk of life and it seems that the time for the onset of susceptibility is placed between PND28 and PND35. However, MDMA induced neurotoxicity at an earlier postnatal age in rats pretreated concomitantly with the dopamine precursor L-DOPA. MDMA was not either neurotoxic in adult rats with a previous dopaminergic lesion but the neurotoxicity was reinstated in the lesioned animals when MDMA was given in combination with L-DOPA.
By using the MAO-B inhibitor, L-deprenyl, Sprague and Nichols (1995b)![]() prevented MDMA-induced neurotoxicity and blocked the formation of thiobarbituric reactive substances, often viewed as an index of enhanced lipid peroxidation. According to their results and other numerous studies focused on the mechanism of MDMA-induced neurotoxicity, these authors suggested that a possible sequence of events leading to 5-HT axon terminal degeneration after MDMA could be the following: 1) acute 5-HT depletion from serotonergic neurons (Sprague et al., 1994
prevented MDMA-induced neurotoxicity and blocked the formation of thiobarbituric reactive substances, often viewed as an index of enhanced lipid peroxidation. According to their results and other numerous studies focused on the mechanism of MDMA-induced neurotoxicity, these authors suggested that a possible sequence of events leading to 5-HT axon terminal degeneration after MDMA could be the following: 1) acute 5-HT depletion from serotonergic neurons (Sprague et al., 1994![]() ) and acute increase in dopamine release (Nash, 1990
) and acute increase in dopamine release (Nash, 1990![]() ; Nash and Brodkin, 1991
; Nash and Brodkin, 1991![]() ). 2) Stimulation of 5-HT2A receptors, by the released 5-HT, responsible for an increased dopamine synthesis necessary to support MDMA-induced transmitter efflux (Huang and Nichols, 1993
). 2) Stimulation of 5-HT2A receptors, by the released 5-HT, responsible for an increased dopamine synthesis necessary to support MDMA-induced transmitter efflux (Huang and Nichols, 1993![]() ; Koch and Galloway, 1997
; Koch and Galloway, 1997![]() ; Nash et al., 1990
; Nash et al., 1990![]() ; Schmidt et al., 1992a
; Schmidt et al., 1992a![]() ). 3) Transport by the 5-HT carrier of extracellular dopamine into the depleted serotonergic terminal (Faraj et al., 1994
). 3) Transport by the 5-HT carrier of extracellular dopamine into the depleted serotonergic terminal (Faraj et al., 1994![]() ; Schmidt and Lovenberg, 1985
; Schmidt and Lovenberg, 1985![]() ). 4) Deamination of dopamine by MAO-B inside the 5-HT terminal, generating not only DOPAC but also hydrogen peroxide and possibly other oxidative species responsible for lipid peroxidation and final axon degeneration (Sprague and Nichols, 1995b
). 4) Deamination of dopamine by MAO-B inside the 5-HT terminal, generating not only DOPAC but also hydrogen peroxide and possibly other oxidative species responsible for lipid peroxidation and final axon degeneration (Sprague and Nichols, 1995b![]() ).
).
Serotonergic and dopaminergic neurotransmitter systems undergo a significant maturation during the postnatal development of the central nervous system. As MDMA-induced 5-HT and dopamine release is carrier mediated (Koch and Galloway, 1997![]() ; Nash and Brodkin, 1991
; Nash and Brodkin, 1991![]() ; Schmidt, 1987
; Schmidt, 1987![]() ), their respective uptake systems may not be present on 5-HT and dopamine axons; however, this seems unlikely as it has been demonstrated that both uptake systems exhibit a rapid proliferation from PND0 to PND14 (Kirksey and Slotkin, 1979
), their respective uptake systems may not be present on 5-HT and dopamine axons; however, this seems unlikely as it has been demonstrated that both uptake systems exhibit a rapid proliferation from PND0 to PND14 (Kirksey and Slotkin, 1979![]() ). 5-HT2 receptors seem to play also an important role in the maintenance of
MDMA-induced dopamine release and serotonergic toxicity. It has been shown that 5-HT2 receptor agonists potentiate MDMA-induced dopamine release and serotonergic neurotoxicity in adult rats (Gudelsky et al., 1994
). 5-HT2 receptors seem to play also an important role in the maintenance of
MDMA-induced dopamine release and serotonergic toxicity. It has been shown that 5-HT2 receptor agonists potentiate MDMA-induced dopamine release and serotonergic neurotoxicity in adult rats (Gudelsky et al., 1994![]() ) although blockade of these receptors prevents the long-term deficits produced by MDMA (Schmidt et al., 1990a
) although blockade of these receptors prevents the long-term deficits produced by MDMA (Schmidt et al., 1990a![]() ,b
,b![]() ). The lack of sensitivity of rat pups to the long-term brain 5-HT depletion may then indicate that 5-HT2 receptors are not entirely functional at early postnatal ages. Yet, it is known that, in the rat, cells expressing 5-HT2 receptors reach a peak at about the end of the second postnatal week and this is followed by a regressive process until approximately PND28 (Bruinink et al., 1983
). The lack of sensitivity of rat pups to the long-term brain 5-HT depletion may then indicate that 5-HT2 receptors are not entirely functional at early postnatal ages. Yet, it is known that, in the rat, cells expressing 5-HT2 receptors reach a peak at about the end of the second postnatal week and this is followed by a regressive process until approximately PND28 (Bruinink et al., 1983![]() ; Morilak and Ciaranello, 1993
; Morilak and Ciaranello, 1993![]() ; Roth et al., 1991
; Roth et al., 1991![]() ). Consistent with this ontogeny, we found in our study that rats treated at PND21 with the selective 5-HT2 receptor agonist, R-DOI (0.5 mg/kg s.c.), showed a high number of head twitches (not shown), an effect mediated by 5-HT2 receptor stimulation (e.g., Zifa and Fillion, 1992
). Consistent with this ontogeny, we found in our study that rats treated at PND21 with the selective 5-HT2 receptor agonist, R-DOI (0.5 mg/kg s.c.), showed a high number of head twitches (not shown), an effect mediated by 5-HT2 receptor stimulation (e.g., Zifa and Fillion, 1992![]() ), suggesting that 5-HT2 receptors were already functional at this age. Despite this, the combination of R-DOI and MDMA at PND21 neither produced a significant decrease in 5-HT transporter density in the frontal cortex and in the hippocampus nor caused a significant deficit of 5-HT and 5-HIAA in any of the brain regions examined. In consequence, the lack of neurotoxicity after MDMA exposure, at least at PND21, is not apparently due to low 5-HT2 receptor density or functioning.
), suggesting that 5-HT2 receptors were already functional at this age. Despite this, the combination of R-DOI and MDMA at PND21 neither produced a significant decrease in 5-HT transporter density in the frontal cortex and in the hippocampus nor caused a significant deficit of 5-HT and 5-HIAA in any of the brain regions examined. In consequence, the lack of neurotoxicity after MDMA exposure, at least at PND21, is not apparently due to low 5-HT2 receptor density or functioning.
It has been reported (Johnson and Nichols, 1991![]() ) that the combination of the nonneurotoxic serotonin releaser 5-methoxy-6-methyl-2-aminoindan with the nonvesicular dopamine releaser, amphetamine, produces long-term serotonergic deficits similar to those induced by MDMA. However, increased dopamine synthesis and
release after the combination of the selective 5-HT2 receptor agonist R-DOI (at the same dose used in our study) with amphetamine is not sufficient to induce serotonergic deficits (Huang and Nichols, 1993
) that the combination of the nonneurotoxic serotonin releaser 5-methoxy-6-methyl-2-aminoindan with the nonvesicular dopamine releaser, amphetamine, produces long-term serotonergic deficits similar to those induced by MDMA. However, increased dopamine synthesis and
release after the combination of the selective 5-HT2 receptor agonist R-DOI (at the same dose used in our study) with amphetamine is not sufficient to induce serotonergic deficits (Huang and Nichols, 1993![]() ). These authors suggested that it is probably necessary a previous 5-HT depletion from the neuron to render the serotonergic terminal vulnerable to the toxic action of dopamine, as increased dopamine synthesis and efflux cannot explain by itself the neurotoxic effects induced by MDMA. Since acute MDMA already depletes 5-HT from the terminal at the age of 10 days (Broening et al., 1994
). These authors suggested that it is probably necessary a previous 5-HT depletion from the neuron to render the serotonergic terminal vulnerable to the toxic action of dopamine, as increased dopamine synthesis and efflux cannot explain by itself the neurotoxic effects induced by MDMA. Since acute MDMA already depletes 5-HT from the terminal at the age of 10 days (Broening et al., 1994![]() ) and 5-HT2 receptors seem to be functional at PND21, there must be other reason to explain why the long-term neurotoxic effects of MDMA are not observed until a time between PND28 and PND35.
) and 5-HT2 receptors seem to be functional at PND21, there must be other reason to explain why the long-term neurotoxic effects of MDMA are not observed until a time between PND28 and PND35.
Numerous authors have suggested that MDMA-induced neurotoxicity depends on its ability to release dopamine through a carrier-mediated mechanism (e.g., White et al., 1996![]() ). Furthermore, there appears to exist a linear correlation between the acute increase of extracellular dopamine and the extent of serotonergic toxicity induced by MDMA (Nash and Nichols, 1991
). Furthermore, there appears to exist a linear correlation between the acute increase of extracellular dopamine and the extent of serotonergic toxicity induced by MDMA (Nash and Nichols, 1991![]() ). Even though studies on the functional ontogeny of dopaminergic neurons have demonstrated that they are able to initiate or conduct action potentials at approximately PND8 (Cheronis et al., 1979
). Even though studies on the functional ontogeny of dopaminergic neurons have demonstrated that they are able to initiate or conduct action potentials at approximately PND8 (Cheronis et al., 1979![]() ; Erinoff and Heller, 1978
; Erinoff and Heller, 1978![]() ), adult levels of striatal dopamine are not reached until approximately PND30 (Nomura et al., 1976
), adult levels of striatal dopamine are not reached until approximately PND30 (Nomura et al., 1976![]() ). Another possibility to explain the lack of sensitivity to MDMA in newborn rats is that extracellular dopamine concentrations are not high enough to get into the serotonergic terminal, producing the oxidative oxygen species responsible for the neurotoxicity. Supporting this hypothesis, we found that the combined treatment with the dopamine precursor, L-DOPA, and MDMA at PND21 significantly reduced the concentrations of 5-HT and 5-HIAA in the frontal cortex and in the hippocampus and also decreased significantly the number of [3H]paroxetine-labeled 5-HT transporters in these two brain regions. It could be also supposed that norepinephrine and not dopamine was involved in 5-HT depletion, as norepinephrine concentration is higher in the hippocampus than in the striatum whereas the striatum, having the highest dopamine concentration, was less affected than the hippocampus. However, it has been suggested (Schmidt et al., 1991a
). Another possibility to explain the lack of sensitivity to MDMA in newborn rats is that extracellular dopamine concentrations are not high enough to get into the serotonergic terminal, producing the oxidative oxygen species responsible for the neurotoxicity. Supporting this hypothesis, we found that the combined treatment with the dopamine precursor, L-DOPA, and MDMA at PND21 significantly reduced the concentrations of 5-HT and 5-HIAA in the frontal cortex and in the hippocampus and also decreased significantly the number of [3H]paroxetine-labeled 5-HT transporters in these two brain regions. It could be also supposed that norepinephrine and not dopamine was involved in 5-HT depletion, as norepinephrine concentration is higher in the hippocampus than in the striatum whereas the striatum, having the highest dopamine concentration, was less affected than the hippocampus. However, it has been suggested (Schmidt et al., 1991a![]() ), that such considerations only become relevant by assuming a terminal-terminal interaction between the 5-HT and dopaminergic systems, a structural aspect still unresolved at the time of analyzing the mechanism(s) of action of MDMA. Further, desipramine should prevent the uptake of 6-hydroxydopamine into the noradrenergic nerve terminal (e.g., review by Kostrzewa and Jacobowitz, 1974
), that such considerations only become relevant by assuming a terminal-terminal interaction between the 5-HT and dopaminergic systems, a structural aspect still unresolved at the time of analyzing the mechanism(s) of action of MDMA. Further, desipramine should prevent the uptake of 6-hydroxydopamine into the noradrenergic nerve terminal (e.g., review by Kostrzewa and Jacobowitz, 1974![]() ), so a partial reduction of 5-HT content in animals previously lesioned with 6-hydroxydopamine should be expected if norepinephrine were involved in MDMA-induced neurotoxicity. Decreased
5-HT concentrations and 5-HT transporter density were only found when MDMA was given to 6-hydroxydopamine-lesioned rats in combination with L-DOPA. These results support an important role for dopamine in the mechanism of neurotoxicity induced by MDMA. It should be noted that Perry et al. (1995)
), so a partial reduction of 5-HT content in animals previously lesioned with 6-hydroxydopamine should be expected if norepinephrine were involved in MDMA-induced neurotoxicity. Decreased
5-HT concentrations and 5-HT transporter density were only found when MDMA was given to 6-hydroxydopamine-lesioned rats in combination with L-DOPA. These results support an important role for dopamine in the mechanism of neurotoxicity induced by MDMA. It should be noted that Perry et al. (1995)![]() , found lasting serotonergic deficits after p-chloroamphetamine in adult rats neonatally treated with 6-hydroxydopamine. Although the neurotoxicity induced by p-chloroamphetamine is apparently similar to that seen after MDMA, the mechanisms are not at all identical and drugs such as dizocilpine or deprenyl are able to prevent the long-term neurotoxicity of MDMA but not that of p-chloroamphetamine (Colado and Green, 1994
, found lasting serotonergic deficits after p-chloroamphetamine in adult rats neonatally treated with 6-hydroxydopamine. Although the neurotoxicity induced by p-chloroamphetamine is apparently similar to that seen after MDMA, the mechanisms are not at all identical and drugs such as dizocilpine or deprenyl are able to prevent the long-term neurotoxicity of MDMA but not that of p-chloroamphetamine (Colado and Green, 1994![]() ; Sprague et al., 1996
; Sprague et al., 1996![]() ).
).
The serotonergic deficits found in PND21 rats after the combined treatment of L-DOPA and MDMA were not as dramatic as those found in adult animals. Colado et al. (1997)![]() , using the same rat strain and a higher MDMA dose (40 mg/kg), did not find increased formation of thiobarbituric acid reacting substances in neonates (PND7-10). These authors suggested that the lack of sensitivity of neonates to the neurotoxic effects of MDMA and other related amphetamines could be due to a much higher capacity of neonates to scavenge free radicals. This possibility, along with the much lower intracellular concentration of dopamine in immature rats, could be determinant for the lack of neurotoxicity of MDMA at early postnatal ages.
, using the same rat strain and a higher MDMA dose (40 mg/kg), did not find increased formation of thiobarbituric acid reacting substances in neonates (PND7-10). These authors suggested that the lack of sensitivity of neonates to the neurotoxic effects of MDMA and other related amphetamines could be due to a much higher capacity of neonates to scavenge free radicals. This possibility, along with the much lower intracellular concentration of dopamine in immature rats, could be determinant for the lack of neurotoxicity of MDMA at early postnatal ages.
It has been also suggested that the sensitivity of the immature rat to MDMA-induced hyperthermia develops concomitantly with the
sensitivity of the immature rat to the long-term neurotoxic effects of MDMA on the serotonergic system (Broening et al., 1995![]() ). As indicated (see "Methods"), temperature was measured by reinserting the probes from time to time. Although it is known that colonic temperature of rats can be slightly raised by handling (Gordon, 1990
). As indicated (see "Methods"), temperature was measured by reinserting the probes from time to time. Although it is known that colonic temperature of rats can be slightly raised by handling (Gordon, 1990![]() ), this factor should not probably exert an influence on the results as experimental conditions were identical for all animal groups. MDMA caused a significant and long-lasting hyperthermia when administered at PND21 without producing 1 wk later any significant deficit in 5-HT
content or in 5-HT transporter density. Furthermore, the combination of R-DOI and MDMA produced in the rat pups a similar or slightly higher hyperthermia than the combination of L-DOPA and MDMA. However, only the latter treatment was able to induce a lasting reduction in the density of 5-HT transporters and 5-HT content in the frontal cortex and in the hippocampus. According to our results, it appears that the hyperthermia induced by MDMA is not sufficient to produce the lasting neurotoxic effects on the serotonergic system at least in the 21-day-old rats used in these experiments.
), this factor should not probably exert an influence on the results as experimental conditions were identical for all animal groups. MDMA caused a significant and long-lasting hyperthermia when administered at PND21 without producing 1 wk later any significant deficit in 5-HT
content or in 5-HT transporter density. Furthermore, the combination of R-DOI and MDMA produced in the rat pups a similar or slightly higher hyperthermia than the combination of L-DOPA and MDMA. However, only the latter treatment was able to induce a lasting reduction in the density of 5-HT transporters and 5-HT content in the frontal cortex and in the hippocampus. According to our results, it appears that the hyperthermia induced by MDMA is not sufficient to produce the lasting neurotoxic effects on the serotonergic system at least in the 21-day-old rats used in these experiments.
In summary, our results appear to indicate that the lack of neurotoxicity to MDMA exposure at early postnatal ages is probably due to an insufficient concentration of dopamine in the rat brain, and argue for a certain threshold of dopamine release to observe a serotonergic deficit. It is consequently proposed that an already developed dopaminergic system is critical for the long-term expression of serotonergic neurotoxicity in rats by MDMA.
FOOTNOTESAccepted for publication May 8, 1998.
Received for publication June 16, 1997.
1 This work was supported by EEC (BIO4 CT96-0752) and CICYT (970315).
Send reprint requests to: Dr. Joaquín Del Río, Department of Pharmacology, University of Navarra Medical School, c/Irunlarrea 1, 31008 Pamplona, Spain.
ABBREVIATIONSDOPAC, dihydroxyphenylacetic acid; E, embryonic day; HVA, homovanillic acid; 5-HT, 5-hydroxytryptamine, serotonin; 5-HIAA, 5-hydroxyindoleacetic acid; L-DOPA, L-3,4-dihydroxyphenylalanine; MDMA, 3,4-methylenedioxymethamphetamine; PND, postnatal day; R-DOI, (R)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane.
REFERENCES
- Aguirre N, Galbete JL, Lasheras B and Del Río J (1995) Methylenedioxymethamphetamine induces opposite changes in central pre- and postsynaptic 5-HT1A receptors in rats. Eur J Pharmacol 281: 101-105
- Battaglia G, Yeh SY, O'Hearn E, Molliver ME, Kuhar MJ and De Souza EB (1987) 3,4-Methylenedioxymethamphetamine and 3,4-Methylenedioxyamphetamine destroy serotonin terminals in rat brain: quantification of neurodegeneration by measurement of [3H]paroxetine-labeled serotonin uptake sites. J Pharmacol Exp Ther 242: 911-916
- Broening HW, Bacon L and Slikker W, Jr (1994) Age modulates the long-term but not the acute effects of the serotonergic neurotoxicant 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther 271: 285-293
- Broening HW, Bowyer JF and Slikker W, Jr (1995) Age-dependent sensitivity of rats to the long-term effects of the serotonergic neurotoxin (±)-3,4-methylenedioxymethamphetamine (MDMA) correlates with the magnitude of the MDMA-induced thermal response. J Pharmacol Exp Ther 275: 325-333
- Bruinink A, Lichtensteiger W and Schlumpf M (1983) Pre- and postnatal ontogeny and characterization of dopaminergic D2, serotonergic S2, and spirodecanone binding sites in rat forebrain. J Neurochem 40: 1227-1236
- Cadet JL, Ladenheim B, Hirata H, Rothman RB, Ali S, Carlson E, Epstein C and Moran TH (1995) Superoxide radicals mediate the biochemical effects of methylenedioxymethamphetamine (MDMA): Evidence from using CuZn-superoxide dismutase transgenic mice. Synapse 21: 169-176
- Cheronis JC, Erinoff L, Heller A and Hoffmann PC (1979) Pharmacological analysis of the functional ontogeny of the nigrostriatal dopaminergic neurons. Brain Res 169: 545-560
- Colado MI and Green AR (1994) A study on the mechanism of MDMA ("Ecstasy")-induced neurotoxicity of 5-HT neurones using chlormethiazole, dizocilpine and other protective compounds. Br J Pharmacol 111: 131-136
- Colado MI and Green AR (1995) The spin trap reagent -phenil-N-tertbutyl nitrone prevents "ecstasy"-induced neurodegeneration of 5-hydroxytryptamine neurons. Eur J Pharmacol 28: 343-346
- Colado MI, O'Shea E, Granados R, Misra A, Murray TK and Green AR (1997) A study of the neurotoxic effect of MDMA ("ecstasy") on 5-HT neurones in the brains of mothers and neonates following administration of the drug during pregnancy. Br J Pharmacol 121: 827-833
- Erinoff L and Heller A (1978) Functional ontogeny of nigrostriatal neurons. Brain Res 142: 566-569
- Faraj BA, Olkowski ZL and Jackson RT (1994) Active [3H]-dopamine uptake by human lymphocites: Correlates with serotonin transporter activity. Pharmacol 48: 320-327
- Gordon CJ (1990) Thermal biology of the laboratory rat. Physiol Behav 47: 962-991
- Gudelsky GA and Nash JF (1996) Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: Implications for serotonin-dopamine interactions. J Neurochem 66: 243-249
- Gudelsky GA and Yamamoto BK (1994) MDMA increases the extracellular concentration of 2,3-dihydroxybenzoic acid in the striatum: Evidence for increased hydroxyl radical formation. Soc Neurosci Abstr 24: 419.1
- Gudelsky GA, Yamamoto BK and Nash JF (1994) Potentiation of 3,4-methylenedioxymethamphetamine-induced dopamine release and serotonin neurotoxicity by 5-HT2 agonists. Eur J Pharmacol 264: 325-330
- Gudelsky GA (1996) Effect of ascorbate and cysteine on the 3,4-methylenedioxymethamphetamine-induced depletion of brain serotonin. J Neural Transm 103: 1397-1404
- Huang X and Nichols DE (1993) 5-HT2 receptor-mediated potenciation of dopamine synthesis and central serotonergic deficits. Eur J Pharmacol 238: 291-296
- Johnson MP and Nichols DE (1991) Combined administration of a non neurotoxic 3,4-methylenedioxymethamphetamine analogue with amphetamine produces serotonin neurotoxicity in rats. Neuropharmacology 30: 819-822
- Kirksey DF and Slotkin TA (1979) Concomitant development of [3H]-dopamine and [3H]-5-hydroxytryptamine uptake systems in rat brain regions. Br J Pharmacol 67: 387-391
- Koch S and Galloway MP (1997) MDMA induced dopamine release in vivo: role of endogenous serotonin. J Neural Transm 104: 135-146
- Kostrzewa RM and Jacobowitz DM (1974) Pharmacological actions of 6-hydroxydopamine. Pharmacol Rev 26: 199-288
- Malberg JE, Sabol KE and Seiden LS (1996) Co-administration of MDMA with drugs that protect against MDMA neurotoxicity produces different effects on body temperature in the rat. J Pharmacol Exp Ther 278: 258-267
- Marcusson JO, Bergström M, Eriksson K and Ross SB (1988) Characterization of [3H]paroxetine binding in rat brain. J Neurochem 50: 1783-1790
- Morilak DA and Ciaranello RD (1993) Ontogeny of 5-Hydroxytryptamine2 receptor immunoreactivity in the developing rat brain. Neuroscience 55: 869-880
- Nash JF and Brodkin J (1991) Microdialysis studies on 3,4-methylenedioxymethamphetamine-induced dopamine release: effect of dopamine uptake inhibitors. J Pharmacol Exp Ther 259: 820-825
- Nash JF and Nichols DE (1991) Microdialysis studies of 3,4-methyelendioxymethamphetamine and structurally related analogues. Eur J Pharmacol 200: 53-58
- Nash JF, Meltzer HY and Gudelsky GA (1990) Effect of 3,4-methylenedioxymethamphetamine on 3,4-dihydroxyphenylalanine accumulation in the striatum and nucleus accumbens. J Neurochem 54: 1062-1067
- Nash JF (1990) Ketanserin pretreatment attenuates MDMA-induced dopamine release in the striatum as measured by in vivo microdialysis. Life Sci 47: 2401-2408
- Nomura Y, Naitoh F and Segawa T (1976) Regional changes in monoamine content and uptake of the rat brain during postnatal development. Brain Res 101: 305-315
- O'Hearn E, Battaglia G, De Souza EB, Kuhar MJ and Molliver ME (1988) Methylenedioxyamphetamine (MDA) and Methylenedioxymethamphetamine (MDMA) cause selective ablation of serotonergic axon terminals in forebrain: Inmunocytochemical evidence for neurotoxicity. J Neurosci 8: 2788-2803
- Pérez-Otaño I, Herrero MT, Oset C, De Ceballos ML, Luquin MR, Obeso JA and Del Río J (1991) Extensive loss of brain dopamine and serotonin induced by chronic administration of MPTP in the marmoset. Brain Res 567: 127-132
- Perry KW, Kostrzewa RM and Fuller RW (1995) Persistence of long-lasting serotonin depletion by p-chloroamphetamine in rat brain after 6-hydroxydopamine lesioning of dopamine neurons. Biochem Pharmacol 50: 1305-1307
- Roth BL, Hamblin MW and Ciaranello RD (1991) Developmental regulation of 5-HT2 and 5-HT1C mRNA and receptor levels. Dev Brain Res 58: 51-58
- Schmidt CJ and Lovenberg W (1985) In vitro demonstration of DA uptake by neostriatal serotonergic neurons of the rat. Neurosci Lett 59: 9-14
- Schmidt CJ and Taylor VL (1987) Depression of rat brain tryptophan hydroxylase following the acute administration of methylenedioxymethamphetamine. Biochem Pharmacol 36: 4095-4102
- Schmidt CJ, Abbate GM, Black CK and Taylor VL (1990a) Selective 5-hydroxytryptamine2 receptor antagonist protect against the neurotoxicity of methylenedioxymethamphetamine in rats. J Pharmacol Exp Ther 255: 478-483
- Schmidt CJ, Black CK and Taylor VL (1990b) Antagonism of the neurotoxicity due to a single administration of methylenedioxymethamphetamine. Eur J Pharmacol 181: 59-70
- Schmidt CJ, Black CK and Taylor VL (1991a) L-DOPA potentiation of the serotonergic deficits due to a single administration of 3,4-methylenedioxymethamphetamine, p-chloroamphetamine or methamphetamine to rats. Eur J Pharmacol 203: 41-49
- Schmidt CJ, Black CK, Taylor VL, Fadayel GM, Humphreys TM, Nieduzak TR and Sorensen SM (1992a) The 5-HT2 receptor antagonist, MDL 28,133A, disrupts the serotonergic-dopaminergic interaction mediating the neurochemical effects of 3,4-methylenedioxymethamphetamine. Eur J Pharmacol 220: 151-159
- Schmidt CJ, Fadayel GM, Sullivan CK and Taylor VL (1992b) 5-HT2 receptors exert a state-dependent regulation of dopaminergic function: studies with MDL 100,907 and the amphetamine analogue 3,4-methylenedioxymethamphetamine. Eur J Pharmacol 223: 65-74
- Schmidt CJ, Sullivan CK and Fadayel GM (1994) Blockade of striatal 5-hydroxytryptamine2 receptors reduces the increase in extracellular concentrations of dopamine produced by the amphetamine analogue 3,4-methylenedioxymethamphetamine. J Neurochem 62: 1382-1389
- Schmidt CJ, Taylor VL, Abbate GM and Nieduzak TR (1991b) 5-HT2 antagonists stereoselectively prevent the neurotoxicity of 3,4-methylenedioxymethamphetamine by blocking the acute stimulation of dopamine synthesis: reversal by L-Dopa. J Pharmacol Exp Ther 256: 230-235
- Schmidt CJ (1987) Neurotoxicity of the psychedelic amphetamine methylenedioxymethamphetamine. J Pharmacol Exp Ther 240: 1-7
- Slikker W, Jr, Ali SF, Scallet AC, Frith CH, Newport GD and Bailey JR (1988) Neurochemical and neurohistological alterations in the rat and monkey produced by orally administered methylenedioxymethamphetamine (MDMA). Toxicol Appl Pharmacol 94: 448-457
- Sprague JE and Nichols DE (1995a) Inhibition of MAO-B protects against MDMA-induced neurotoxicity in the striatum. Psychopharmacology 118: 357-359
- Sprague JE and Nichols DE (1995b) The Monoamine Oxidase-B inhibitor L-Deprenyl protects against 3,4-methylenedioxymethamphetamine-induced lipid peroxidation and long-term serotonergic deficits. J Pharmacol Exp Ther 273: 667-673
- Sprague JE, Huang X, Kanthasamy A and Nichols DE (1994) Attenuation of 3,4-methylenedioxymethamphetamine (MDMA) induced neurotoxicity with the serotonin precursors tryptophan and 5-hydroxytryptophan. Life Sci 55: 1193-1198
- Sprague JE, Johnson MP, Schmidt CJ and Nichols DE (1996) Studies on the mechanism of p-chloroamphetamine neurotoxicity. Biochem Pharmacol 52: 1271-1277
- St. Omer VEV, Ali SF, Holson RR, Duhart HM, Scalzo FM and Slikker W, Jr (1991) Behavioral and neurochemical effects of prenatal methylenedioxymethamphetamine (MDMA) exposure in rats. Neurobehav Toxicol Teratol 13: 13-20
- Stone DM, Johnson M, Hanson GR and Gibb JW (1988) Role of endogenous dopamine in the central serotonergic deficits induced by 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther 247: 79-87
- Stone DM, Stahl DC, Hanson GR and Gibb JW (1986) The effects of 3,4-Methylenedioxymethamphetamine (MDMA) and methylenedioxyamphetamine (MDA) on monoaminergic systems in the rat brain. Eur J Pharmacol 128: 41-48
- White SR, Obradovic T, Imel KM and Wheaton MJ (1996) The effects of methylenedioxymethamphetamine (MDMA, "ecstasy") on monoaminergic neurotransmission in the central nervous system. Progr Neurobiol 49: 455-479
- Zifa E and Fillion G (1992) 5-Hydroxytryptamine receptors. Pharmacol Rev 44: 401-458
0022-3565/98/2863-1159$03.00/0
THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS
Copyright © 1998 by The American Society for Pharmacology and Experimental Therapeutics