
By Erowid
(with thanks to Bob for supplying the data)
Understanding Drug-Drug interactions and contraindications is complex and involves a number of factors. Two of the primary elements in the current understanding of drug activity are the P450 liver enzymes and the MAO (Mono-Amine Oxidase) enzyme found throughout the body. The following tables include information about various P450 enzyme subtypes and their interaction with various chemicals.
Contents:
Author, Title, Year, Page.. ?
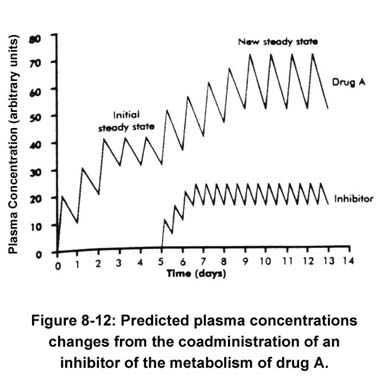 Concentration rises to a new steady state consistent with a change in its clearance. The time to achieve the new steady state is greater than the time to achieve the initial steady state, because the half-life is now prolonged from its original value. The full effect of an inhibitory interaction may not be realized until the inhibitor also reaches a steady state, because the degree of inhibition will also depend on the concentration of the inhibitor (Houston 1994; von Moltke et al. 1994, 1995).
Concentration rises to a new steady state consistent with a change in its clearance. The time to achieve the new steady state is greater than the time to achieve the initial steady state, because the half-life is now prolonged from its original value. The full effect of an inhibitory interaction may not be realized until the inhibitor also reaches a steady state, because the degree of inhibition will also depend on the concentration of the inhibitor (Houston 1994; von Moltke et al. 1994, 1995).
Drug interactions are graded phenomena. The degree of interaction depends on the concentration of interacting drugs and, therefore, on the dose and timing of administration. Drug interactions are most likely to be detected when therapy with an interacting drug is initiated or discontinued. The clinical significance will depend on the particular drugs involved, the physiological state of the patient, the Presence of concurrent illness, and other factors. Drugs with a narrow concentration range over which therapeutic effects are present without incurring toxicity are more likely to be involved in clinically significant drug interactions. These drugs include theophylline, some anticonvulsants and antiarrhythmics (Table 8-1)[not shown here].
When selecting a specific drug from a class of drugs to treat mental illness efficacy, safety, cost, and a history of tense are pertinent considerations. The introduction of the SSRIs has emphasized the critical importance of also considering potential drug interactions (Brosen 1996). These antidepressants have been shown in vitro and in vivo to be potent inhibitors of some cytochrome P450 isoenzumes (Crewe et al. 1992; Nemeroff et al. 1996; von Moltke et al. 1994, 1995). The most thoroughly studied reaction is the competitive inhibition of CYP2D6. Pharmacokinetic studies in healthy volunteers have provided a rank order of the potency for increasing the plasma concentration of model substrates. Case reports in patients have confirmed the existence of some interactions, and many others remain theoretical possibilities. Table 8-4 provides an overall ranking of the cytochrome P450 inhibitory potential of the newer antidepressants based on both in vitro and in vivo data. The rational selection of an antidepressant should include consideration of its potential enzyme inhibition when therapy is to be combined with substrates listed in Table 8-1, which may be inhibited by the specific antidepressant. Reviews (Harvey and Preskorn 1996; Nemeroff et al. 1996) describe the specific in vivo reports in more detail.
 The selection of a drug based on its cytochrome P450 inhibitory potential should not be limited to the newer antidepressants (Table 8-4). Combining any two drugs that are substrates for the same enzyme increases the likelihood of competitive enzyme inhibition. All of the substrates listed in Table 8-1 are potential inhibitors. For example, nortriptyline, desipramine, and thioridazine are potent inhibitors of CYP2D6. In vitro methods using microsomal incubations to predict in vivo interactions have appeared and are based on accepted pharmacokinetic principles (Gillette 1971; Houston 1994; von Moltke et al. 1994, 1995). These screening techniques are now used extensively in the pharmaceutical industry in drug development.
The selection of a drug based on its cytochrome P450 inhibitory potential should not be limited to the newer antidepressants (Table 8-4). Combining any two drugs that are substrates for the same enzyme increases the likelihood of competitive enzyme inhibition. All of the substrates listed in Table 8-1 are potential inhibitors. For example, nortriptyline, desipramine, and thioridazine are potent inhibitors of CYP2D6. In vitro methods using microsomal incubations to predict in vivo interactions have appeared and are based on accepted pharmacokinetic principles (Gillette 1971; Houston 1994; von Moltke et al. 1994, 1995). These screening techniques are now used extensively in the pharmaceutical industry in drug development.
From PDH: Psychotropic Drug Handbook, pages 454-459
Table 8-1 lists the various drugs metabolized by CYP450 isoenzymes l Al, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4, as well as the drugs that inhibit these enzymes. Many of these potential drug interactions have not been studied; however, they are predictable from a theoretical basis. Thus, although a theoretical drug interaction may be obvious according to Table 8?1, it may not be described in this chapter either because it is of minor significance or because it has not been studied (DeVane 1994; Gelenberg 1995; Gonzalez and Idle 1994; Harvey and Preskorn 1996a, 1996b; Ishaid et al. 1996; Slaughter and Edwards 1995; Wrighton and Stevens 1992). Concurrently administered drugs metabolized by the same P450 enzymes can demonstrate competitive inhibition of one another's metabolism.
Antipsychotics
Contents:
- Clinically Important Drug-Drug Interactions:
The Importance of Hepatic Cytochrom P-450 Isozyme Activity
- PDH: Antidepressant Pharmacokinetic Parameters
- Concentrations & Antidepressant P450 Interactions
- Potential CYP450 drug interactions for U.S.-marketed drugs
| Table 21-9. Clinically Important Drug-Drug Interactions: The Importance of Hepatic Cytochrome P-450 Isozyme Activity From Pharmacokinetic Mechanisms of Drug Interactions page 313 |
||||
|---|---|---|---|---|
| Cytochrome | Polymorphism | Substrate | Inhibitors | Inducers |
| 1A2 | Possible |
Caffeine, Phenacetin, tacrine, theophylline |
Cimetidine, Fluvoxamine, Quinalones |
Cigarette Smoke |
| 2C9 | Yes |
Diclofenac, ibuprofen, naproxen, phenytoin, piroxicam, S-warfarin, tolbutamide |
Azoles |
Rifampin |
| 2D6 | Yes |
Captropril, codeine, debrisoquine, Dextromethorphan, ecainide, flecainide, fluoxetine, norfluoxetine, haloperidol, imipramine, nortriptyline, paroxetine, risperidone, thioridazine |
Cimetidine |
Rifampin(?) |
| 2C19 | Yes |
Diazepam, imipramine, omeprazole, propranolol, S-mephenytoin |
Rifampin |
|
| 2E1 | No |
Dapsone, theophylline? |
Isoniazid |
|
| 3A3/3A4 | Possible |
Astemizole, cisapride, cyclosporine, erythromycin, itraconazole, lidocaine, loratadine, lovastatin, midazolam, quinidine, tacrolimus, taxol, terfenadine, verapamil |
Azoles |
Carbamazepine |
Coadministration of drugs that are metabolized via the same primary cytochrome isozyme have
the potential of precipitating a clinically important drug-drug interaction. |
||||
| PDH: Psychotropic Drug Handbook, page 143 | ||||
|---|---|---|---|---|
| Table 2-1. Antidepressant Pharmacokinetic Parameters | ||||
| Drug | Bioavailability (%) | Free Drug (%) | Volume of Distribution (L/kg) |
Half-life (hours) |
Amitriptyline | 30-60 | 3-15 | 6.4-36 | 9-46 |
Amoxapine | 46-82 | -- | -- | 8.8-14 |
Bupropion | >90 | 20 | 27-63 | 9.6-20.9 |
Clomipramine | 36-62 | 2-10 | 9-25 | 15-62 |
Desipramine | 33-51 | 8-27 | 24-60 | 12-28 |
Doxepin | 13-45 | 15-32 | 9-33 | 8-25 |
Fluoxetine | ~72 | 5 | 12-42 | 26-220 |
Fluvoxamine | >90 | 33 | >5 | 13-19 |
Imipramine | 22-77 | 4-37 | 9.3-23 | 6-28 |
Maprotiline | 79-87 | 12 | 16-32 | 27-50 |
Nortriptyline | 46-70 | 7-13 | 15-23 | 18-56 |
Paroxetine | >90 | 5 | 3-28 | 7-37 |
Protriptyline | 75-90 | 6-10 | 15-31 | 54-198 |
Sertraline | -- | 5 | -- | ~25 |
Trazodone | 70-90 | 5-11 | .8-1.5 | 6.3-13 |
Trimipramine | 18-63 | 3-7 | 17-48 | 16-40 |
Venlafaxine | 13 | 70 | 6 | 4 |
Source: DeVane 1992; DeVane and Jarecke 1992; Ellingrod amd Perry 1995. | ||||
Author, Title, Year, Page.. ?
Concentration rises to a new steady state consistent with a change in its clearance. The time to achieve the new steady state is greater than the time to achieve the initial steady state, because the half-life is now prolonged from its original value. The full effect of an inhibitory interaction may not be realized until the inhibitor also reaches a steady state, because the degree of inhibition will also depend on the concentration of the inhibitor (Houston 1994; von Moltke et al. 1994, 1995).Drug interactions are graded phenomena. The degree of interaction depends on the concentration of interacting drugs and, therefore, on the dose and timing of administration. Drug interactions are most likely to be detected when therapy with an interacting drug is initiated or discontinued. The clinical significance will depend on the particular drugs involved, the physiological state of the patient, the Presence of concurrent illness, and other factors. Drugs with a narrow concentration range over which therapeutic effects are present without incurring toxicity are more likely to be involved in clinically significant drug interactions. These drugs include theophylline, some anticonvulsants and antiarrhythmics (Table 8-1)[not shown here].
When selecting a specific drug from a class of drugs to treat mental illness efficacy, safety, cost, and a history of tense are pertinent considerations. The introduction of the SSRIs has emphasized the critical importance of also considering potential drug interactions (Brosen 1996). These antidepressants have been shown in vitro and in vivo to be potent inhibitors of some cytochrome P450 isoenzumes (Crewe et al. 1992; Nemeroff et al. 1996; von Moltke et al. 1994, 1995). The most thoroughly studied reaction is the competitive inhibition of CYP2D6. Pharmacokinetic studies in healthy volunteers have provided a rank order of the potency for increasing the plasma concentration of model substrates. Case reports in patients have confirmed the existence of some interactions, and many others remain theoretical possibilities. Table 8-4 provides an overall ranking of the cytochrome P450 inhibitory potential of the newer antidepressants based on both in vitro and in vivo data. The rational selection of an antidepressant should include consideration of its potential enzyme inhibition when therapy is to be combined with substrates listed in Table 8-1, which may be inhibited by the specific antidepressant. Reviews (Harvey and Preskorn 1996; Nemeroff et al. 1996) describe the specific in vivo reports in more detail.
The selection of a drug based on its cytochrome P450 inhibitory potential should not be limited to the newer antidepressants (Table 8-4). Combining any two drugs that are substrates for the same enzyme increases the likelihood of competitive enzyme inhibition. All of the substrates listed in Table 8-1 are potential inhibitors. For example, nortriptyline, desipramine, and thioridazine are potent inhibitors of CYP2D6. In vitro methods using microsomal incubations to predict in vivo interactions have appeared and are based on accepted pharmacokinetic principles (Gillette 1971; Houston 1994; von Moltke et al. 1994, 1995). These screening techniques are now used extensively in the pharmaceutical industry in drug development. From PDH: Psychotropic Drug Handbook, pages 454-459
Table 8-1 lists the various drugs metabolized by CYP450 isoenzymes l Al, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4, as well as the drugs that inhibit these enzymes. Many of these potential drug interactions have not been studied; however, they are predictable from a theoretical basis. Thus, although a theoretical drug interaction may be obvious according to Table 8?1, it may not be described in this chapter either because it is of minor significance or because it has not been studied (DeVane 1994; Gelenberg 1995; Gonzalez and Idle 1994; Harvey and Preskorn 1996a, 1996b; Ishaid et al. 1996; Slaughter and Edwards 1995; Wrighton and Stevens 1992). Concurrently administered drugs metabolized by the same P450 enzymes can demonstrate competitive inhibition of one another's metabolism.
Antipsychotics
- Guanethidine (Major Severity)
Chlorpromazine doses greater than 100 mg/day have been found to reverse the antihypertensive effects of guanethidine Oanowsky et al. 1973). Haloperidol 6-9 mg/day and thiothixene 60 mg/day were found to produce a similar effect (Janowsky et al. 1973). Chlorpromazine blocks the presynaptic neuronal amine uptake pump, thereby denying guanethidine its site of action. Thus, the use of guanethidine is contraindicated in patients receiving antipsychotic medications.
- Anticholinergics (Moderate Severity)
Controlled studies have indicated that the beneficial effects of both chlorpromazine and haloperidol, especially in regard to cognitive function and social behavior, are reversed by anticholinergic drugs (Singh and Kay 1979). The authors have suggested that the central anticholinergic effects are responsible for his antagonism. However, another study has shown that anticholinergics can reduce plasma levels of oral chlorpromazine by enhancing metabolism in the gut (Rivera?Calimlin et al. 1976). Because of these potential interactions, it is recommended that antiparkinsonian (anticholinergic) drug doses he kept to a minimum when treating extrapyramidal reactions secondary to antipsychotic medications.
| Table 8-1 Potential CYP450 drug interactions for U.S.-marketed drugs | |||
|---|---|---|---|
| P450 isoenzyme | Metabolized by | Inhibited by | Induced by |
1A2 |
acetominophen, amitriptyline, antipyrine,caffeine, clomipramine, clozapine, enoxacin, fluvoxamine, haloperidol, imipramine, olanzapine, ondansetron, phenacetin, propranolol, tacrine, theophylline, R(-)warfarin, verapamil |
cimetidine, erythromycin, fluroquinolones, grapefruit juice (naringenin), methoxsalen, paroxetine, sertraline |
charbroiled meat,cruciferous vegetables (e.g., broccoli, cabbage,sprouts), omeprazole, tobacco |
2A6 | coumarin | ||
286 | cyclophosphamide | ||
2C8 | arachidonic acid, paclitaxel, retinoic acid,warfarin | ||
2C9 | cyclophosphamide, diclofenac, hexobarbital, ibuprofen, mefanamic acid, naproxen, phenytoin, piroxicam, tenoxicam, thiotepa, tolbutamide, TCAs, torsemide, S(-)warfarin | sertraline | rifampin, barbiturates |
2C19 | clomipramine, diazepam, hexobarbital,imipramine, lansoprozole, mephenytoin, mephobarbital, moclobemide,omeprazole, proguarnil, -propranolol |
felbamate, fluoxetine, fluvoxamine, fluconazole, sertraline | |
2D6 | Antiarrhythmics | alfentanil, amiodarone | rifampin |
Antipsychotics | cimetidine, fentanil | ||
Beta-Blockers | fluvoxamine, norfluoxetine | ||
Opiates [Erowid Note] | propoxyphene, quinidine | ||
TCAs | sertraline, yohimbine | ||
SSRIs | |||
Miscellaneous antidepressants | &nsbp; | ||
Miscellaneous | |||
2E1 |
acetominophen, chlorzoxazone, ethanol, enflurane, halothane | disulfiram | |
3A4 |
Antiarrhythmics |
cimetidine, ciprofloxacin, anhydroerythromycin, fluvoxamine,grapefruit juice (naringenin), itraconazole, ketoconazole, nefazodone, norfluoxetine,sertraline, troleandomycin | barbiturates, carbamazepine,glucocorticoids,phenytoin, rifampin,rifabutin |
Antidepressants | |||
Benzodiazepines | |||
Calcium channel blockers | |||
Nonsedating antihistamines | |||
Miscellaneous | |||
Note. SSRIs = selective serotonin reuptake inhibitors; TCAs = tricyclic antidepressants. | |||
Notes #
- Note About Dextromethorphan (DXM):
Dextromethorphan was originally listed here in the Opiates category. Although DXM was once thought to be an opioid, since the mid 1990s research has shown that it does not share the standard mu-opioid receptor activity.Although this issue is somewhat complicated, one of the receptor types DXM acts on (the 'Sigma' receptors) was formerly called an "opioid" receptor. DXM is now known to work as a Sigma agonist and as an NMDA antagonist, although there continue to be confusing crossovers with opiates and opioids. See: Kamei 1998: Mechanisms of Central Antitussives, and Redwine 2003: Effects of NMDA receptor antagonists on acute mu-opioid analgesia in the rat.